Overview
FragPipe-Analyst
FragPipe-Analyst is an easy-to-use, interactive web application developed to perform differential expression analysis with “one click” and to visualize quantitative proteomic datasets analyzed using FragPipe computational platform. It is compatible with the LFQ-MBR, TMT, and DIA quantification workflows in FragPipe. FragPipe-Analyst is based on the original LFQ-Analyst code.
Features
- Differential expression analysis
- Enrichment analysis (GO/Pathways)
- Imputation (optional)
- Data visualization
- PCA
- Sample correlation
- Heatmaps
- Missing values inspection
- Sample coverage
- Protein intensity plots for selected protein(s)
- Imputation effect evaluation
Example Results
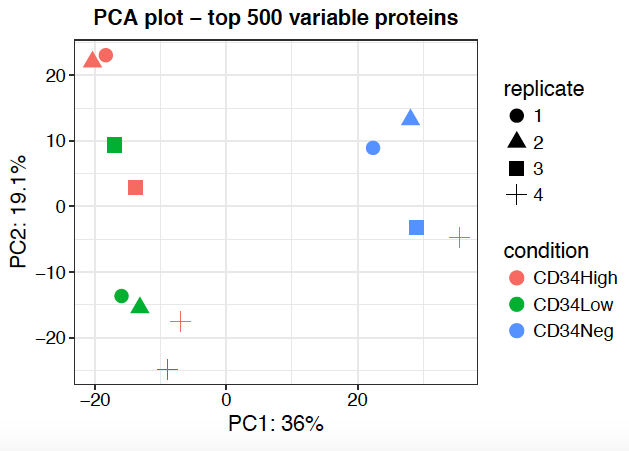
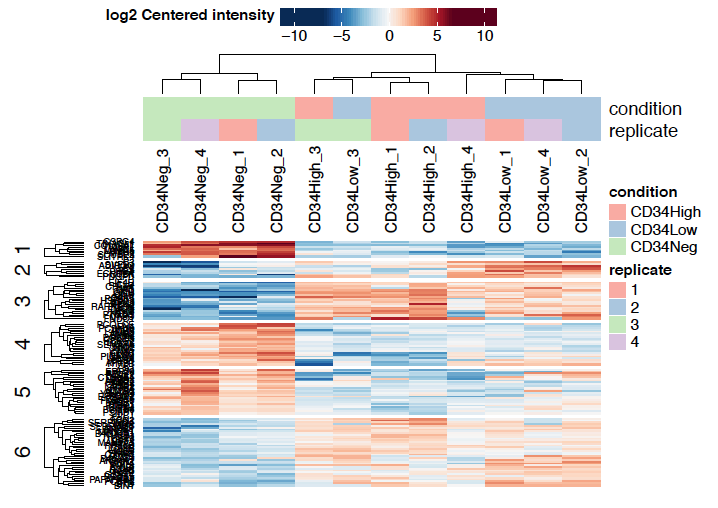
Sidebar tabs
- Analysis: perform your own analysis
- Documentation: Learn more about how to use FragPipe-Analyst
Support
- Questions/Suggestions/Bug reports: Ask us in our GitHub forum .
- Documentation/Tutorials: Learn more here .
- Servers: Our production (stable) server is at https://fragpipe-analyst.org/ but we also provide our latest dev server http://fragpipe-analyst.nesvilab.org/ with most recent updates and bug fixes.
Getting Started
Quick Start
- Choose the type of experiment you performed. Currently, DDA-based LFQ (MS1-based or spectral count), TMT, and DIA are supported. Users could also choose to perform analysis at the protein or peptide levels.
-
For DDA LFQ:
- For protein: Upload combined_protein.tsv generated by IonQuant in FragPipe
- For peptide: Upload combined_peptide.tsv generated by IonQuant in FragPipe
- Upload experiment_annotation.tsv file. Edit the template file generated by FragPipe; Check here for details.
- Select quantification method (MS1-based Intensity or MaxLFQ Intensity, or spectral counts).
-
For TMT:
- For protein or gene: Upload gene-level report [abundance/ratio]_gene_[normalization].tsv or protein-level report [abundance/ratio]_protein_[normalization].tsv generated by TMT-Integrator (TMT-I) in FragPipe (we recommend abundance_gene_MD.tsv file).
- For peptide: Upload peptide-level report [abundance/ratio]_peptide_[normalization].tsv generated by TMT-Integrator (TMT-I) in FragPipe (we recommend abundance_peptide_MD.tsv file).
- For site: Upload single-site report [abundance/ratio]_single-site_[normalization].tsv generated by TMT-Integrator (TMT-I) in FragPipe (we recommend abundance_single-site_MD.tsv file).
- Upload experiment_annotation.tsv file. Edit the template file generated by FragPipe; Check here for details.
-
For DIA:
- For protein: Upload protein group (PG) matrix ( report.pg_matrix.tsv ) generated by DIA-NN in FragPipe
- For peptide: Upload peptide-level report ( abundance_peptide_[MS1/MS2quant]_[normalization].tsv ) generated by by FragPipe (we recommend abundance_peptide_MS2quant_Norm.tsv file).
- For site: Upload single site report ( [abundance/ratio]_single-site_[normalization].tsv ) generated by FragPipe
- Upload experiment_annotation.tsv file. Check here for details.
- Optional: Adjust the missing value filter, normalization, missing value imputation, p-value cut-off, the log2 fold change cut-off, FDR correction method in the Advanced Options . Note that the missing value imputation method is set by default to “Perseus-like” for DDA LFQ and DIA, and to “No imputation” for TMT.
- Press 'Run'
- Hint: Check the Documentation tab for a detailed explanation of inputs, advanced options and outputs
Results Table
Filter Options
Filtering your identification result based on predeefined filter conditions, and/or changing the sliders of each condition below
Subset Results Table
Number of samples present
Filtered Results Table
Documentation
Need help?
- Read our documentation and tutorial here .
- Report issues and ask questions here .
- FragPipe-Analyst is open-source! You are more than welcome to contribute .
- Learn more about our FragPipe here .
- The user manual of original LFQ-Analyst can be accessed here .
Contact Us
For any feedback or question regarding FragPipe-Analyst, please contact the Proteomics & Integrative Bioinformatics Lab (P.I. Alexey Nesvizhskii; University of Michigan):
- Yi Hsiao: yihsiao@umich.edu
- Professor Alexey Nesvizhskii: nesvi@med.umich.edu
News and Updates
- 03-11-2024: Preprint of FragPipe-Analyst is available in bioRxiv.
- 12-02-2022: FragPipe-Analyst is first released for beta testing.
- 07-13-2022: FragPipe-Analyst is first created.